 Publications
Publications
New Ms: CaMKII activation in Barth syndrome
Liu, X. et al., Circulation in press
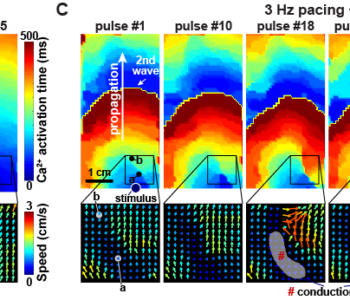
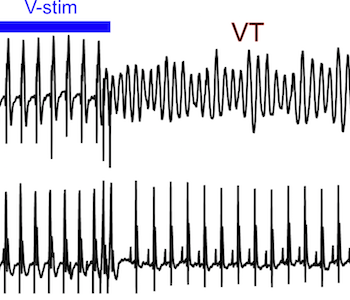
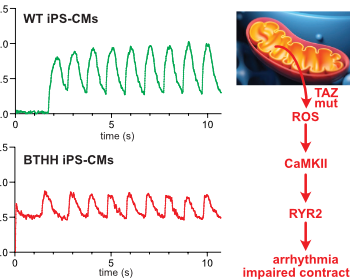
Barth syndrome cardiomyocytes from human iPSCs and mice have abnormal calcium handling that is partially due to activation of CaMKII. Barth syndrome mice had increased vulnerability to arrhythmia, perhaps reflecting the abnormal calcium handling.