 Publications
Publications
Manuscript accepted — Reference map of cardiac transcription factor…
Full Text available here.
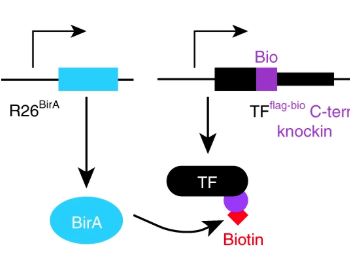
Mapping transcription factors (TFs) occupancy is essential for understanding transcriptional programs. Here Akerberg, Gu, and colleagues use biotinylated knockin alleles of key cardiac TFs (GATA4, NKX2-5, MEF2A, MEF2C, SRF, TBX5, TEAD1) to map their genome-wide occupancy in the fetal and adult mouse heart, providing insight into the cardiac transcriptional regulatory network.